

A Creationist Perspective of Beneficial Mutations in Bacteria
Abstract
Mutations alter the nucleotide sequence of the DNA. They may affect the organism’s phenotype, which can play a key role in bacterial adaptation and transformation to changing environments. Some of these mutations even appear to be beneficial to the organism. However, creationists have tended to offer an inconsistent or incomplete perspective of “beneficial mutations” within a creation framework. This includes the frequent denial that mutations can ever provide a beneficial phenotype, and the concept that “beneficial mutations” are merely an evolutionist exaggeration.
Keywords: Mutation, Hypermutable, Evolution, Adaptive mutation, Natural selection, DNA, Bacteria, Environment, Generations, Survival, Starvation, Temperature, Antibiotic resistance, Nylon degradation, Horizontal gene transfer
This paper was originally published in the Proceedings of the Sixth International Conference on Creationism, pp. 73–86 (2008) and is reproduced here with the permission of the Creation Science Fellowship of Pittsburgh (www.csfpittsburgh.org).
In bacteria, a wide range of mutations can be shown to provide a beneficial phenotype to the cell. These benefits are often of sufficient phenotypic affect that they can undergo strong positive selection. But the benefits are generally temporary and limited. Some common examples of beneficial mutations are those involved in bacterial antibiotic resistance. These mutations potentially enable the bacterium to survive exposure to various antibiotics, but the resistance results from loss or reduction of pre-existing activities such as enzymatic, regulatory, or transport systems. Bacteria also can undergo adaptive mutation; a phenomenon used by bacteria to survive very specific stressful conditions. The exact mechanism is controversial because some results suggest a directed mutation specifically enabling adaptation to the environment but at a mutation rate higher than random mutations would produce. Various mutations have also been found that enable bacteria to survive temporary exposure to high temperatures or starvation. Such mutations usually involve loss of certain sigma factors, reduction of DNA repair, or loss of specific regulatory controls. Other examples include several subpopulations of mutant strains of bacteria obtained over a period of up to 20,000 generations. These mutants have a greater “fitness” than the wild-type strain. However, analysis showed that most contained deletion mutations in various genes.
Each of these examples, as well as numerous others, involves certain environmental conditions that make these mutations phenotypically beneficial. However, these mutations frequently eliminate or reduce pre-existing cellular systems and functions. This has been referred to as antagonistic pleiotropy; meaning the cell experiences a trade-off where a temporary benefit for surviving one environmental condition is provided at the expense of systems used for other environments. If the environmental conditions change, the mutation usually becomes less beneficial and perhaps even detrimental. Hence, these mutations do not provide a genetic mechanism that accounts for the origin of biological systems or functions. Rather, they require the prior existence of the targeted cellular systems. As such, beneficial mutations of bacteria fit concisely within a creation model where (a) biological systems and functions were fully formed at creation, (b) subsequent mutations can provide conditional benefits that enable the organism to survive harsh post-Fall conditions even though the mutation is generally degenerative, and (c) most bacteria need the ability to rapidly adapt to ever changing environments and food sources.
Introduction
All DNA appears to have the potential for mutation. A mutation can be simply defined as a change in the DNA nucleotide sequence compared to a wild-type sequence.1 These mutations often alter the organism’s phenotype, which can play a key role in biological adaptation and transformation. Many mutations clearly are deleterious to the organism, probably even lethal. Other types of mutations appear to provide some, albeit, temporary benefit. Creationists have tended to offer inconsistent evaluations of such “beneficial” mutations. Morris2 categorized beneficial mutations as a prediction of an evolutionary model, implying their occurrence is inconsistent with a creation model. Other creationists have denied that mutations ever provide a beneficial phenotype.
However, a wide range of mutations can be shown to provide a beneficial phenotype to an organism. These benefits are often of sufficient phenotypic affect that they can undergo strong positive selection. Such benefits, though, are often temporary and limited.
Bacteria have become a popular system for studying mutations and alleged evolutionary transformations.
Bacteria are asexual microorganisms that can rapidly reproduce. While it would appear that bacterial offspring are effectively a clone of the parent, this genetic homogeneity would reduce a bacterial population’s ability to quickly respond to changing environments. Instead, bacteria have numerous mechanisms for introducing genetic variation into a growing population.3 These mechanisms, combined with their rapid reproduction and large population sizes, enable bacteria to quickly and effectively adapt to a variety of environmental changes.
Because of these features, bacteria have become a popular system for studying mutations and alleged evolutionary transformations.4, 5 A variety of mutations have been studied in bacteria. Many are clearly deleterious, but examples of advantageous or “beneficial” mutations can also be found.
Many published examples of beneficial mutations are actually proposed historical reconstructions of possible past mutational events. While such reconstructions may accurately represent a mutation’s history, it is still a speculated history. In fact, most reconstructions also are based more upon evolutionary supposition than genetic plausibility. Instead, in this paper we will focus upon those mutations where both the wild-type and mutant strain can be empirically compared side-by-side, and the mutation history is clearly documented. Also, some beneficial mutations have resulted from directed genetic engineering. Such engineered alterations do not necessarily represent naturally occurring mutations, rather they are illustrations of theoretical genetic possibilities. In fact, they are genetic possibilities that can result from a guiding intelligence.
Examples of Beneficial Mutations
Mutations during multiple generations
Lenski et al.6 determined that after 10,000 generations in the same cultivation conditions, a population of Escherichia coli contained a diversity of mutant strains. Each of these mutants had an increased adaptation to the cultivation condition, manifesting itself in a 50% greater relative fitness compared to the parental wildtype. Thus, the mutations appeared to be beneficial and were positively selected.
Subsequent genetic analysis of some of these E. coli mutants found that they possessed insertion sequences (IS elements; a small segment of DNA that can insert into numerous sites of the chromosome). These IS elements were indigenous to the chromosome, and their activity did not depend on horizontal transfer from neighboring cells. Movement of these elements produced various insertional mutations in the E. coli chromosome.7 In fact, these types of mutations appear to be the primary function of these IS elements.8 These insertional mutations either create “knockout mutations”, disrupting gene function or genetic activity at the point of insertion, or they may carry a promoter or other regulatory segments that activate adjacent genes.9 One specific IS disruption of gene activity in some of the mutants was the loss of the ribose operon.10
No specific explanation has yet been offered of how IS-mediated disruptions are advantageous, but “antagonistic pleiotropy” has been proposed.11 This pleiotropy suggests that a mutation can be beneficial in one type of environment and deleterious for a different environment. Thus, the cell seems to readily sacrifice a system not essential to a given environment, if it in some way helps adaptation to that environment.
After 20,000 generations, additional E. coli mutant strains were isolated that also appeared to be positively selected. Some of these strains had a reduced expression of flagella-encoding (flg) genes.12 Since the parental wild-type was non-motile, maintenance of the unused flagella-encoding genes was likely an unnecessary burden in the cultivation conditions. This provides another example of genomic truncation leading to antagonistic pleiotropy.
A second group of mutants from the same study probably involved a mutation in the regulatory control of spoT.13 This mutation can subsequently result in reduction of ppGpp concentration, a molecule involved in stress response.14 Reduction of ppGpp concentration, in turn, may increase tRNA and rRNA expression.15 Increased RNA can contribute to increased growth rate of the mutants.16
All the E. coli mutants detected within 20,000 generations appear to fit the description of antagonistic pleiotrops. The beneficial mutations are very environmentally specific, and a change in this environment often negates the benefit of the mutation. When repeatedly cultivated in a constant environment, it is not surprising that an organism would reduce its genome of some unused genes and functions. Mutants of E. coli obtained after 20,000 generations at 37°C were less “fit” than the wild-type strain when cultivated at either 20°C or 42°C.17 Other E. coli mutants obtained after 20,000 generations in medium where glucose was their sole catabolite tended to lose the ability to catabolize other carbohydrates.18 Such a reduction can be beneficially selected only as long as the organism remains in that constant environment. Ultimately, the genetic effect of these mutations is a loss of a function useful for one type of environment as a trade-off for adaptation to a different environment.
Stress Survival
Adaptive mutation
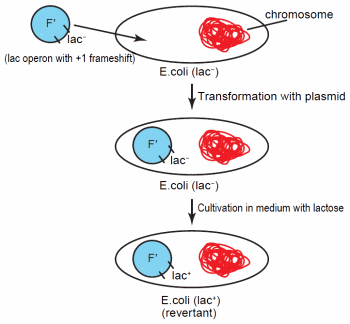
Figure 1. Transformation of Escherichia coli with the F' plasmid containing the lac operon. The operon possesses a + 1 frameshift, so it is unable to express genes for lactose catabolism. Following specific cultivation conditions with lactose containing medium, the lactose operon on some of the plasmids revert to lac+.
In a broad context, adaptive mutation has been defined as that collection of growth-independent mutations that enhance the cell’s survival and growth when confronted with stressful and growth-limiting environments.19 As such, various examples of beneficial mutations are a form of adaptive mutation. Interestingly, adaptive mutations appear to be mutations that arise specifically in response to the environment.
The initial observed behavior of adaptive mutations suggested they arise from non-random mutations that are directed specifically to adapt to a particular environmental cue or stressor.20, 21 Hence, they were originally termed “directed mutations” because they appeared to be non-randomly generated mutations. However, other mutations often occur concurrently with “directed” mutations that are not directly a result of environmental responses.22
Early work on adaptive mutations involved Lacstrains of E. coli or Salmonella enterica with an F' plasmid containing a copy of the lac operon with a + 1 frameshift (Figure 1). This frameshift renders the operon inactive. While the cells initially remain phenotypically Lac- with this plasmid, after cultivation on medium with lactose as the sole energy source, Lac+ revertants developed at a rate higher than predicted from random mutation.23 In addition, the culture experienced no overall net growth.23
There is some controversy about the nature of adaptive mutations. Are directed mutations specifically arising due to selective pressure, almost in a Lamarckian fashion? Or, are mutations occurring in a more conventional, random manner, and selection processes effectively produce the appearance of “directed” mutations? Focusing on the Lac- mutant experiment described above as the model study system, three models have been proposed for adaptive mutation.
For these Lac+ revertants to occur during a phase of stationary growth (that is, no net growth) requires various recombination enzymes, such as RecA24 and induction of the SOS DNA damage response regulon.25 In particular, the error-prone DNA polymerase IV may have significant involvement. Based on this, one model proposed is the recombination-dependent mutation model (also known as directed mutation).26 This complicated model assumes the mutated lac operon on the plasmid is “leaky,” enabling enough lactose catabolism to provide sufficient energy for a low level of DNA replication (Figure 2). Even though conjugation is not occurring, the conjugal origin on the F' plasmid sustains persistent nicks. When the DNA replication fork encounters these nicks, the fork collapses and creates a double-strand end on the plasmid. This double strand end initiates the dsDNA break repair system in the cell involving the recombination enzyme, RecA. RecA catalyzes invasion of a single DNA strand into this double-strand break, priming DNA synthesis. If the error prone DNA polymerase IV is recruited for the DNA synthesis, an increased number of mutations are potentially introduced in the inactive lac operon on the plasmid. This, in turn, increases the probability of a reversion (-1 frameshift) in lac copy.
A second model is the amplification mutagenesis model. This model suggests that the reversion involves gene duplication of the lac region.27, 28 Like the previous model, this model assumes the lac operon on the plasmid possesses enough lactose catabolic activity that some cells can replicate. In some of these replicating cells, the lac region on the plasmid is duplicated. This model assumes that the reversions do not occur in the non-growing parental cells. Instead, some of the growing cells duplicate the lac region, which possesses some weak lactose catabolizing activity. Unequal recombination increases the number of lac copies per cell, enabling even more growth of these cells. This amplified number of lac operons increases the probability of a reversion (-1 frameshift) in the developing clones. No change in either rate or specificity of mutagenesis occurs. But, this model does require the +1 frameshift mutation to be “leaky” and that lactose be present in the selective media.29
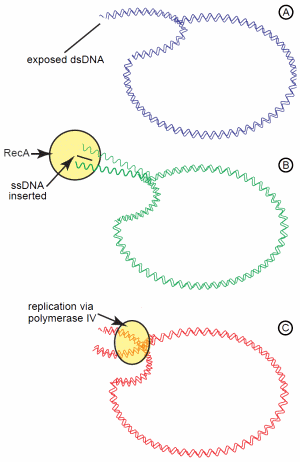
Figure 2. The recombination-dependent mutation model for adaptive mutations of the lactose operon on the F' plasmid. The “leaky” +1 frameshift of the lac operon provides enough energy from lactose catabolism to initiate replication of the plasmid. (A) Persistent nicks in the plasmid cause the replication fork to collapse and leave exposed dsDNA. (B) Exposed dsDNA initiates DNA repair with RecA, which inserts a ssDNA segment into the dsDNA. (C) Replication of the DNA by the low fidelity DNA polymerase IV gives an increased potential of replication errors leading to numerous mutations.
The problem for many researchers was that adaptive mutations did not fit their neo-Darwinian paradigm. Eager to dismiss the concept, many geneticists probably found initial comfort in the third proposed model, the hypermutation model.30 This model claims that the revertants resulted from randomly generated mutations in the Lac- mutants. These Lac- mutants underwent a state of hypermutation, which increases the probability of a reversion (-1 frameshift) of the lac operon on the F' plasmid. The hypermutation model was initially supported by claims of 20 to 50-fold increases of nonselected mutations in the revertants.31 However, Roth et al.32 challenged that achieving the observed number of lac revertants would require the overall mutation rate to be implausibly high. It was also found that only about 10% of the adaptive mutants appear to have been hypermutators.33 Thus, hypermutation does not appear to be required for adaptive mutation within this specific F' model.
Starvation
Helling, Vargas, and Adams34 obtained mutant strains of E. coli following prolonged cultivation under glucose-limited conditions. They found that a stable co-existing population of two different types of mutant strains developed. One mutant possessed a greater rate of glucose uptake v. the wild-type strain. Glucose uptake in the wild-type strain is regulated by a variety of feedback mechanisms, including glucose metabolites. However, this mutant apparently increases glucose uptake by truncating its pathways for glucose catabolism,35 and acetate is a dominant endproduct. Theoretically, this mutant results from a reduction of feedback inhibition, enabling increased glucose uptake.
The second mutant detected in this co-existing population had mutations that reduced the regulatory control of acetyl CoA synthetase, causing over-expression of this enzyme.36 This resulted in an enhanced ability of the mutants to scavenge the exogenous levels of acetate being produced by the other mutant. Thus, together these mutants had a greater survival in the glucose-limited cultivation conditions. However, again, these two mutants are a form of antagonistic pleiotropy. The phenotypic benefit from the mutations was at the expense of a metabolic pathway necessary for more efficient glucose catabolism, and a regulatory system that is necessary for normal genetic control. Also, the second mutant strain is only beneficial in conjunction with the other mutant. Unless that mutant occurs, the acetyl-CoA mutant will not be advantageous.
A different set of bacterial mutants was obtained by cultivating Pseudomonas putida in starvation conditions for a prolonged period in phenol-based minimal medium. An initial population of mutants involved reduced activity of the DNA repair enzyme MutY.37 Oxidation of guanine to 7,8-dihydro-8-oxoguanine is a common type of DNA damage in starving bacterial cells.38 The MutY repair enzyme is involved in repair of this damage. Reduction of this repair activity probably precipitates an increase in C → A transversions.39 As the culture continued to survive the starvation period, the population of transversion mutants was replaced by mutants containing 1-3 bp deletions.40, 39 In addition, prolonged starvation appears to increase the activity of indigenous IS elements in the cells,41, 39 presumably generating an increase of “knock out” mutations as a consequence.
Error-prone DNA polymerase also appears to contribute to survival of E. coli under starvation conditions.42 Loss or reduction of mismatch repair in E. coli43 and Bacillus subtilis44 significantly increases their survival in aging cultures. In fact, decreased DNA repair fidelity and activity is typically associated with nutritionally stressed bacteria.45 Presumably, the decrease of DNA repair enables increased rates of mutagenesis (hypermutation).8 Increased mutation rates provide an increased possibility of a mutation beneficial to survival in a given environment. Thus, hypermutation has been associated with bacterial adaptation to a variety of stressful conditions.8
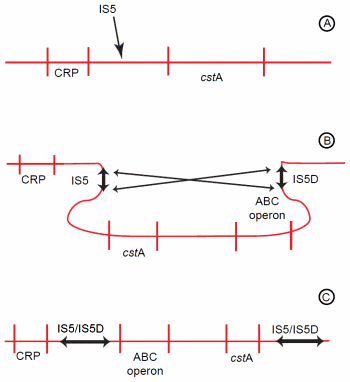
Figure 3. Genomic rearrangement resulting from activity of an indigenous IS element. (A) IS5 inserts between the CRP-binding site and the cstA gene. (B) IS5 then undergoes inversion with IS5D, which is already located upstream of an ABC-type transporter operon. (C) The inversion causes the regions between IS5 and IS5D to invert. Hence, the ABC-type transporter operon is now positioned near the CRP-binding site and becomes activated.
Severe stress, such as stationary-phase aging of bacterial cells, also selects for a mutation in transcription factors. A mutation in the sigma factor, sS, reduces the factor’s regulatory activity, which appears to increase survival of stationary-phase aging cells.46 However, the mutated sS was most advantageous at a high pH, and was detrimental at a low pH.47 Also, a mutation resulting in a three-base-pair deletion of the leucine-responsive regulatory protein (Lrp) eliminates its DNA-binding activity.48 Lrp activates amino acid anabolism and represses amino acid catabolism.49 This mutant form of the lrp gene appears to increase survival of aging cells in stationary-phase.50 Presumably the advantage of either phenotype in aging cells is the loss or reduction of transcription regulation by these factors, although the exact molecular benefit for less transcription control is still speculative.51 Also, inactivation of the tricarboxylic acid (TCA) cycle enzyme, aconitase, enhances survival of cells in the stationary-phase.52
Another mutation of E. coli facilitated amino acid catabolism under starvation conditions, enabling the mutant to outcompete the parental wild-type.53 This increased catabolism resulted from a genomic rearrangement (Figure 3). The first step of this rearrangement was insertion of an indigenous IS5 element between the promoter and a CRP-binding site (catabolite regulatory protein) of the starvationinducible cstA gene.54 The cstA gene encodes an oligopeptide permease.55 Following its insertion, the IS5 undergoes an inversion with a pre-existing IS5 element located about 60 kb distant.56 This second IS element (IS5D) is located upstream of an operon encoding a high affinity ABC-type transporter.57 As a result of the inversion, the ABC-type transport operon is repositioned to close proximity of the CRP-binding site, and cstA is moved away from its regulatory region.56 The cstA gene is inactivated, but the ABC-type transporter operon is activated.57
The adaptive advantage of these mutations under starvation conditions appears to result from an enhanced ability to scavenge amino acids, specifically aspartate and glutamate.57 Simultaneously inactivating oligopeptide uptake and activating increased amino acid uptake helps to serve this purpose. Also, inactivation of lrp enhances amino acid scavenging, but negatively effects amino acid biosynthesis. Hence, these mutants involve a trade-off of resource utilization where amino acid utilization is enhanced and peptide utilization is depressed.
Each of these mutant strains has an antagonistic pleiotrophy characteristic. An existing system is traded for an altered phenotype that is better suited to survive the specific stressful environment. Regulation is reduced to enable overexpression. DNA repair and DNA polymerase fidelity are reduced to enable increased mutation rates (increasing the probability of a “beneficial” mutation). A gene is inactivated by a process that concurrently activates a silent gene. Such trade-offs provide a temporary benefit to the bacterium, increasing its chances of surviving specific starvation conditions. However, these mutations do not account for the origin of the silenced genes, as their prior existence is essential for the mutation to be beneficial.
Temperature Stress
Following cultivation of E. coli for 2,000 generations at 41.5ºC, various groups of thermal adapted mutants were isolated.58 Several of these mutants contained at least one duplication near the 2.85 Mb position on the chromosome. In addition, a 12 kb deletion was detected in one line of mutants. These duplication and deletion events were not detected in the parental strain following cultivation for 2,000 generations at 37°C.59
Duplication of certain genes or groups of genes has been suggested as an adaptive strategy for bacteria. Hence, certain environmental conditions seem to favor bacteria with specific genes duplicated.60, 61 This may have provided the organism a temporary increase in gene expression of those duplicated genes, which apparently helped the organism cope with the higher temperature. However, once the duplicated regions no longer provide an advantage, they are likely eliminated by recombination.59
A second study found that after 2,000 generations at 41.5 °C, specific heat-inducible genes were expressed at significantly higher levels that in wild-type cells.62, 63 Not all known heat-inducible genes were affected, and not all lines of the thermal adapted mutants possessed the same level of expression of each gene. While the mutation of regulatory systems presumably was involved, the exact mechanism was not disseminated.
Antibiotic Resistance
When confronted with antimicrobials, such as antibiotics, bacteria frequently will develop resistance to the compound. While this resistance often results from horizontal transfer of pre-existing resistance genes, a significant number also result from point mutations.8, 64 However, as discussed in detail by Anderson,65 these mutations typically reduce or inactivate cellular systems or functions. Thus, the organism develops a resistance to an antibiotic by eliminating certain systems, such as transport proteins, enzymatic activity, and binding affinity. For example, ampicillin resistance can result from an SOS response that halts cell division,66 loss of nitroreductase activity can provide resistance to metronidazole and amoxicillin,67 and kanamycin resistance can result from loss of a specific transport protein.68
Like many of the other beneficial mutations just discussed, mutations providing antibiotic resistance are a trade-off. In fact, they offer a very clear example of antagonistic pleiotropy. The organism sacrifices an existing system with the trade-off that the antibiotic is no longer activated, transported, bound, etc.
Nylon Degradation
Nylon 6 is a synthetic polymer consisting of more than 100 units of 6-aminohexanoic acid. Other forms of cyclic and non-cyclic nylon oligomers are formed as part of nylon 6 synthesis. Because nylon is not a natural occurring molecule, bacteria would not have been exposed to this polymer until the 20th century. The recent appearance of nylon degrading bacteria presents an interesting demonstration of bacterial ability to adapt to an ever changing environment and substrate. It has also lead to a few highly exaggerated claims regarding bacterial evolution.69
At the phenotypic level, the appearance of nylon degrading bacteria would seem to involve “evolution” of new enzymes and transport systems. However, further molecular analysis of the bacterial transformation reveals mutations resulting in degeneration of pre-existing systems. The most studied of the nylon degrading bacteria is Arthrobacter sp. K172 (formerly Flavobacterium sp.70). This bacterium employs three enzymes for nylon degradation, EI (NylA), EII (NylB), and EIII (NylC), which are found on the plasmid, pOAD2.71, 72
EI and EIII (also NylC in Agromyces sp.) have been initially characterized.73, 72 They apparently hydrolyze the cyclic forms of some nylons, which provides a linear substrate for EII. However, no detailed analysis of the mutational changes of EI or EIII has yet been performed.
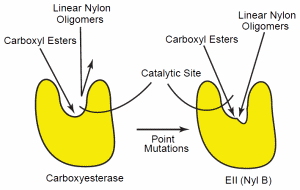
Figure 4. Confirmational change of the carboxyesterase. The esterase (left) can hydrolyze carboxy esters, but the confirmation specificity of the enzyme’s catalytic site does not allow hydrolysis of other polymers, such as nylon. Point mutations in the enzymes’ gene can cause a conformational alteration of the enzyme’s catalytic site so that specificity is reduced (right). This reduced specificity now allows the enzyme to hydrolyze a wider variety of oligomers, including the linear polymer, nylon-6.
The mutational changes of EII (6-aminohexanoatedimer hydrolase) have been characterized in detail. This analysis suggests that point mutations in a carboxyesterase gene lead to amino acid substitutions in the enzyme’s catalytic cleft. This altered the enzyme’s substrate specificity sufficiently that it could also hydrolyze linear nylon oligomers.74, 75 Yet, the EII enzyme still possesses the esterase function of the parent esterase. Thus, the mutational alteration results in a reduction of the parent enzyme’s specificity (Figure 4). This enables it to hydrolyze a wider range of oligomers that include nylon oligomers.76
Nonetheless, reduced specificity of a pre-existing enzyme is biochemically degenerative to the enzyme,77, 78 even if it provides a presumed phenotypic benefit. The “beneficial” phenotype of nylon degradation requires the a priori existence of the enzyme and its specificity. Its degeneration is not a mechanism that accounts for the origin of either the enzyme or its specificity.
Also on pOAD2 is a DNA region with a high homology to opp genes.79 These genes are involved in oligopeptide transport. Nylon oligomers have many chemical similarities to oligopeptides, thus genes on this region of the plasmid may be involved in nylon transport into the cell. No analysis of how these genes may have been altered by mutations has yet been preformed. However, it is reasonable to speculate that a pre-existing opp gene or set of genes has been altered sufficiently by mutations so that the transport proteins now have an affinity for nylon in addition to naturally occurring oligopeptides. As with enzymes, reduction of transport protein specificity is biochemically degenerative.
The enzyme and putative transport genes on pOAD2 appear to form a nylon degrading operon.79 As a plasmid based operon, it can be transferred to various bacterial species. Thus, this gives it the potential for widespread distribution in the bacterial world. What is more, the increasing amount of microbial degradation of synthetic material11 may likely involve a similar mutational strategy as found with nylon degradation. This is a testament to the versatility of bacterial adaptation. However, these mutations do not account for the origin of the enzyme or transport protein specificity, merely their degeneration. Thus, this adaptive versatility has imposed limits as well, and this fits well within the types of mutational changes predicted by a creation model.
Horizontal Gene Transfer
Horizontal gene transfer (HGT) is cell-to-cell transfer of genetic material, usually plasmids or transposons. Plasmids are autonomous extrachromosomal segments of DNA. They may posses a variety of genes, including genes for mobilization and antibiotic resistance.
HGT of plasmids merely transfers genetic material from one cell to another. Introduction of specific genes, such as antibiotic resistance genes, via a plasmid can be beneficial for bacterial survival. However, HGT of plasmids does not directly account for the origin of the gene(s) being transferred, merely their movement within the microbial world. The genes’ existence is a priori. This is consistent with a creation model where genes, regulatory sites, and other genetic activity were formed at the initial creation event, and are not solely the result of mutational or recombination events. The transfer of pre-existing genes and other genetic activity does not account for the origin of the transferred activity. Also, unless the plasmid incorporates into the host chromosome, it does not result in a mutation. Therefore, the contribution of plasmid HGT to bacterial adaptation is a separate topic outside the scope of this paper.
A transposon is a segment of DNA that can be transferred from cell-to-cell via HGT. The transposon can be maintained on a plasmid, but typically exists autonomous of plasmids, and merely moves from chromosome to chromosome. Most cellular “transformation” by transposons involves (a) incorporation of new genes into the cell from the transposon, (b) activation of “silent” genes, and/or (c) insertional disruption of genes and regulatory sites on the host’s chromosome. The introduction of new genes into a cell (such as the antibiotic resistant genes typically found on commonly studied transposons) may provide new genetic capability to the transformed cell. However, as with plasmids discussed above, this does not account for the origin of these genes. Their existence is a priori in the biological world.
By analogy, this is like transferring money from the left to the right pocket. The right pocket now has money that it did not posses before, but you are no richer. This type of mechanism cannot account for the origin of the money, merely its transfer among the population. As applied to the biological world, HGT of genes fails to explain their origin, thus it fails to fulfill the genetic requirements for common descent. However, HGT does fit within the creation model where the transfer of existing genes would be predicted as an adaptive design feature.
A promoter on a transposon may also be able to serve as a promoter for a “silent” or promoterless gene.80 While this makes for an interesting genetic transformation, it offers no answer to ultimate origin. Both the gene and the promoter are already present in the biological world. What is more, how would a promoterless gene evolve? An evolutionary origin requires an immediate and viable function for any “evolving” gene. Without immediate functionality, the “evolving” sequence would be subject to selective removal. Rather than evolving independent of a promoter and awaiting the arrival of the transposon’s promoter, these promoterless genes merely lost their original promoter function at some point. Thus, promoters from transposons can serve as a surrogate.
While promoter surrogacy may provide a beneficial phenotype to the bacterium, it does not account for the origin of either the gene or the promoter, nor does it directly involve mutations. As such, it too is outside the scope of the paper. Also, surrogacy is not a mechanism providing the genetic requirements for evolution’s common descent of ancestry, where there must be an accounting of the origin of both the promoters and the promoterless genes. However, such surrogacy is fully consistent with a creation model.
Most relevant to the focus of this paper are transpositional gene disruptions. When inserting into the chromosome of the transformed cell, the transposon may disrupt a previously existing genetic system or function. Transposon mutagenesis normally results in “knock out” mutations. On occasion, transpositional disruptions will inactivate a regulatory system. This can result in a phenotypic change of the host, such as constitutive production of an enzyme81 or activation of a “silenced” gene.82 Even though such disruptions may occasionally result in a “beneficial” phenotype, they are another form of antagonistic pleiotropy. The benefit is achieved at the expense of a pre-existing genetic system. Thus, disruption or “knock out” mutations are fully consistent with the overall genetic degeneration predicted by the creation model. But, these mutations are inconsistent with a mechanism to account for the origin of flight, vision, cognition, or any other biological system required by common evolutionary descent.
Some transposons, known as foldback elements, can undergo specific folding when inserted into the host’s chromosome. This folding can subsequently initiate extensive chromosomal rearrangement.83 However, we are not aware that any foldback elements have been identified in bacteria. In addition, while transposon activity can lead to the formation of new transposons (and even plasmids), the genetic activities of the newly formed transposon originated from pre-existing genes, replication sites, etc.84
Implications of Beneficial Mutations for a Creation Model
As these examples demonstrate, there are numerous forms of mutations that can assist bacteria in adapting to various environmental conditions and dealing with adverse environments. Such mutations can be referred to as “beneficial,” in that they serve a benefit to the organism. However, the benefit is generally temporary and transient. In fact, identifying many “beneficial” mutations as a form of antagonistic pleiotropy descriptively suggests the limited value of the mutations. Unnecessary or unused systems are eliminated in a manner that provides a phenotype capable of better surviving a specific environment. While this trade-off enables survival, it is at the expense of a pre-existing system or function. Typically the benefit is also temporary. Beneficial mutations for one type of environment often do not serve a benefit in a different environment, and perhaps are even detrimental to survival in an altered environment.85, 86
Rigid flexibility of bacterial mutations
Mutation and natural selection in bacteria, such as in the case of adaptive mutations or antibiotic resistance, are often touted as “evolution in action.” As has been shown in the examples given, these mutations do not account for the origin of new systems or functions. Many of the phenotypes result from loss of pre-existing systems. Other phenotypes involve activation of inactive systems, but this activation often involves simultaneous inactivation of another system. As such, they are not examples of mutations originating new cellular systems or activities.
Instead, bacteria give every appearance of having been designed with a sophisticated ability to adapt to a variety of extreme conditions. This should not be unexpected. Bacteria potentially face a wide variety of environmental conditions. Most bacteria need the ability to adapt to ever changing environments and food sources, and have little alternative but to adapt or die. A creating Designer would provide such ability to bacteria. This may also be true for other unicellular organisms such as yeast and protists.
Yet, bacteria also possess a “rigid flexibility.” This flexibility allows bacteria the necessary genetic versatility to adapt and survive in ever-changing environments.87 A creation model would predict that bacteria would be designed for rapid and versatile adaptation. But, this change is limited and specific. Bacteria appear to have been created to serve specific ecological roles, such as biomass conversion and organosubstrate degradation. Thus, any adaptive changes would necessarily be limited so that the bacteria will continue to function in these designed roles. The rigidity of possible changes would also limit bacteria to remain within its created baramin (see Frair for a detailed analysis of created baramins88). This conserving or limiting effect of mutations fits with observable evidence of bacterial changes.
The very concept of antagonistic pleiotropy purposes that the mutation creates an antagonistic situation in the cell. A system needed for one set of environmental parameters is sacrificed in order to better adapt to different parameters. However, a change of these parameters may require the cell to reestablish the sacrificed system, and perhaps sacrifice a different system instead. This balance continues through a variety of environmental conditions and stress situations. Yet, at no point has the bacterial cell developed a new system. It is merely managing the systems it already possesses. Instead, these mutations require the prior existence of the targeted systems. As such, beneficial mutations of bacteria fit concisely within a creation model where biological systems and functions were fully formed at creation. Any subsequent mutations may provide conditional benefits that enable the organism to survive harsh conditions, but the mutations are generally degenerative. Thus, antagonistic pleiotropy fits succinctly within a creation model.
The study of advantageous mutations is important for understanding the power and the limitation of mutations for change in bacterial species. A greater understanding of them may assist in developing creation models of speciation and adaptation in the post-Fall and post-Flood world. It may even assist in a better understanding of the development of bacterial pathogenesis from a creationist perspective.
Adaptive mutations are not used by most organisms
Although the differences between prokaryotes and eukaryotes are widely known, they are rarely differentiated in studies of evolutionary change. The mantra seems to be Jacques Monod’s statement that, “Anything found to be true for E. coli must also be true of elephants”.8 But elephants have choices that bacteria do not. If food becomes scarce, elephants can migrate to areas where food is more plentiful. Although some bacteria are motile, dramatic changes in location are not feasible. If the temperature becomes hostile or the preferred nutrients become scarce, bacteria have three basic options: endospore formation (not possible for all types of bacteria), adapt, or die.
Bacteria have been designed with great genetic diversity that sometimes is only accessible through genetic alterations by mutations. For example, the ebg operon may be part of a complex backup system for the lac operon in E. coli.89 Perhaps it has ultimately degenerated so that it does not serve the original backup function, but certain mutations can reactivate portions of it. Many other bacterial systems also seem to have various “backup” systems.90 Even a complex operation, such as polysaccharide degradation, has very few rate limiting systems.91
There are several genetic and non-genetic factors that distinguish bacteria from humans and animals, and allow the former to be successful in using mutations for adaptation. As Sanford92 states,
For all these reasons, selection in such systems [bacteria] is much more effective, precise, and can have much higher resolution. This means that in bacteria, a much smaller proportion of the genome is near neutral and un-selectable (this is why theorists typically prefer to use microbial examples) (p. 74).
Ploidy and cellularity differ
Humans are diploid, meaning that every cell (except germ cells) contains two complete copies of each chromosome. Bacteria are generally haploid; each cell possesses only one complete copy of its single chromosome. Under laboratory cultivation it is possible for an individual cell to posses multiple replication forks, hence it may be partial diploid; that is, contain more than one copy of portions of the chromosome. But it is unlikely this is a normal state, and cell division naturally begins before two complete chromosomes are formed.
Being haploid is advantageous to the bacteria under conditions where beneficial mutations are generated. There are no dominant or recessive genotypes of a given gene. In humans, beneficial mutations are less frequent since mutations in both chromosomal copies of the gene would likely need to occur for the mutations to result in a phenotypic change.
In addition, because of the multicellular composition of humans, altering the function of a particular tissue or organ would require the beneficial mutation either to occur in multiple cells or the mutated cell would have to replace all existing cells. For bacteria, the adaptive mutations need only to occur in a single cell. Sanford93 states in regards to bacteria, “Perhaps most importantly, every cell is subject to selection, independently, every cell division.” (italics in original) (p. 74). This situation is not true for a multicellular organism. A particular tissue or organ would be selected for as a whole, not at the individual cell level. In addition, tissues and organs do not function in isolation. They are part of organs and organ systems that are interconnected. These organ systems must work together properly to allow an organism to maintain homeostasis. The complexity of multicellular organisms makes the applicability of most types of beneficial mutations found in bacteria unlikely.
In particular, adaptive mutations would not be expected in humans since the mutations would likely occur in somatic cells, which are not inherited by their offspring. It is difficult to imagine how adaptive mutations could occur in germ cells since these cells would not be faced with any of the same selective pressures as the somatic cells. Such a situation would revert back to a Lamarckian type evolution—a position with no genetic basis in humans. What is more, even if the adaptive mutations occurred in germ cells, there is no guarantee that offspring will exhibit the trait caused by the mutation. Two germ cells (that is, sperm and egg) must combine to form the zygote, and it is unlikely they would have the same adaptive mutations. On-the-other-hand, the unicellular nature of the bacteria ensures that the mutation is passed to subsequent generations.
Degree of functionality within the genome differs
An increasing proportion of the DNA within the genomes of humans is being found to be functional (that is, “junk” DNA is not junk).94 This makes it difficult to perceive how humans and animals could utilize many of the types of “beneficial” mutations found in bacteria. There are many sequences known as pyknons throughout the human genome that do not code for proteins but appear to have some function (possibly structural or regulatory).95 Also, transcriptional regions have been found within exons of known human genes.96
This overlapping nature of genes and regulatory functions in human and animal DNA means that a mutation has a distinct possibility of affecting multiple aspects of the genome. Some of those affects may be beneficial, but most would be detrimental. Bacteria need more genomic flexibility for survival, so they are unlikely to have these types of sequences or suffer multiple effects from a single mutation. Mutations in the human genome may have many side effects, whereas those in bacteria are likely to have one specific effect. Beneficial mutations in bacteria are more likely to have a phenotypic impact and undergo stronger positive selection.
Population sizes and generation times differ
Bacterial populations reproduce much faster and produce more offspring than humans. This rapid generation time and large population size means that bacteria can afford the “cost” of selection. When a bacterial population faces adverse environmental conditions 99.999% of the cells may fail to adapt and subsequently die. However, the small percentage that do survive are sufficient for the population to survive. It can rapidly replenish the population back to the original population size. The surviving bacteria may have multiple deleterious mutations throughout their genome (that is, such as a result of being in a hypermutable state), however, they can compensate by reversion or suppression mutations. Humans (and vertebrates) have much longer generation times and much fewer offspring than bacteria. Consequently, an accumulation of multiple mutations will ultimately lead to genetic decay.93 The “cost” of selection becomes more than the population can pay.97
Amount of “noise” differs
In population genetics, noise is a combination of genetic and non-genetic factors that affect the heritability of a trait within the population. Human genomes are more affected by noise than bacterial genomes. For example, the human trait of “fitness” is extremely complex and greatly affected by noise.93 This may be because most contemporary humans do not consistently face life threatening situations where speed or strength become vital attributes. So, fitness levels of humans are not under a strong selective pressure. Thus, fitness in humans has a low heritability factor.93 However, bacterial fitness is frequently under strong selection, as it is a necessary aspect of their constant life-death struggle. Bacteria that are “fit” will be able to adapt and survive, whereas those unable to adapt will die.
Adaptive mutations in humans may lead to cancer
Multiple mutations are characteristic of cancer cells. The number of mutations in cancer cells cannot be sufficiently accounted for based on the spontaneous mutation rate alone, even if these cells have been exposed to carcinogens.98 It has been suggested that possible developing cancer cells are much more like E. coli cells under non-lethal selection.99 The quickly dividing cells would soon need a well-developed blood supply in order to continue their rapid divisions. The blood supply will take time to develop and it is plausible that early cancer cells would have limited nutrients available. Spontaneous mutations in cells that lead to a mutator phenotype may be the first step.100 The mutator phenotype would be similar to the hypermutable state in E. coli wherein mutations are made but not repaired because of a faulty DNA repair system (discussed previously).
Adaptive mutations that allow the cancer cells to grow better under these conditions may occur. It has even been shown that tumor cells with mutations in DNA repair systems have normal mutations rates when growing and higher mutation rates when not growing. This suggests these cells are in a type of hypermutable state when under nonlethal selection.101 Hall102 also points out the problematic nature of adaptive mutations for multicellular organisms, such as humans, and he states,
The machinery that produces adaptive mutations would be very useful in single-cell organisms but might be deleterious in multicell organisms where the requirement for organizing cells into tissues means that programmed growth constraints must be applied to most cells. In multicellular organisms the activation of that adaptive-mutagenesis machinery, which would be advantageous to the individual cell in that it would allow that cell to divide rapidly, would be very disadvantageous to the organism, where the rapidly growing population would be described as a tumor. (p. 5673)
Conclusion
Bacteria frequently develop mutations that enable them to survive and adapt to a variety of environmental conditions. These mutations are generated by many different mechanisms, and provide a wide range of phenotypic modifications. However, most of these mutations can be classified as a form of antagonistic pleiotropy. Some existing systems are sacrificed as a means for surviving certain environments.
Antagonistic pleiotropy is a useful feature of a creation model. Bacteria face a variety of environmental conditions and stressful situations. However, in order to survive, they must contend with any environmental condition that confronts them. Antagonistic pleiotropy provides them genetic mechanisms where they can make specific (and potentially detrimental) genetic changes that will then serve in a particular environment. If the environmental conditions change, the mutation usually becomes less beneficial and perhaps even detrimental. Hence, these mutations do not provide a genetic mechanism that accounts for the origin of biological systems or functions. Rather, they require the prior existence of the targeted cellular systems. As such, beneficial mutations of bacteria fit concisely within a creation model where (a) biological systems and functions were fully formed at creation, (b) subsequent mutations can provide conditional benefits that enable the organism to survive harsh conditions even though the mutation is generally degenerative, and (c) most bacteria need the ability to rapidly adapt to ever changing environments and food sources.

Footnotes
- Snyder, L., and W. Champness, 2003. Molecular genetics of bacteria. Washington, DC: ASM Press.
- Morris, H. M. (ed.), 1985. Scientific creationism. El Cajon, California: Master Books.
- Anderson, K. L., 2003. The complex world of gastrointestinal bacteria. Canadian Journal of Animal Science 83:409–427.
- Lenski, R. E., and M. Travisan, 1994. Dynamics of adaptation and diversification: A 10,000-generation experiment with bacterial populations. Proceedings of the National Academy of Science, USA 91:6808–6814.
- Mortlock, R. P., 1984. Microorganisms as model systems for studying evolution. New York, New York: Plenum Press.
- Lenski, R. E., J. A. Mongold, P. D. Sniegowski, M. Travisano, F. Vasi, P. J. Gerrish, and T. Schmidt, 1998. Evolution of competitive fitness in experimental populations of E. coli: What makes one genotype a better competitor than another? Antonie van Leeuwenhoek 73:35–47.
- Schneider, D., E. Duperchy, E. Coursange, R. E. Lenski, and M. Blot, 2000. Long-term experimental evolution in Escherichia coli. IX. Characterization of IS-mediated mutations and rearrangements. Genetics 156:477–488.
- Anderson, Ref. 3.
- Schneider, D., and R. E. Lenski, 2004. Dynamics of insertion sequence elements during experimental evolution of bacteria. Research in Microbiology 155:319–327.
- Cooper, V. S., D. Schneider, M. Blot, and R. E. Lenski, 2001.Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of E. coli B. Journal of Bacteriology 183:2834–2841.
- Schneider and Lenski, Ref. 9.
- Cooper, T. F., D. E. Rozen, and R. E. Lenski, 2003. Parallel changes in gene expression after 20,000 generations of evolution in E. coli. Proceedings of the National Academy of Sciences, USA 100:1072–1077.
- Cooper et al., Ref. 12.
- Johansson, J., C. Balsalobre, S.-Y. Wang, J. Urbonaviciene, D. J. Jin, B. Sondén, and B. E. Uhlin, 2000. Nucleoid proteins stimulate stringently controlled bacterial promoters: A link between the cAMP-CRP and the (p)ppGpp regulons in Escherichia coli. Cell 102:475–485.
- Barker, M., T. Gaal, and R. L. Gourse, 2001. Mechanism of regulation of transcription initiation by ppGpp. Models for positive control based on properties of RNAP mutants and competition for RNAP. Journal of Molecular Biology 305:673–688.
- Sarubbi, E., K. E. Rudd, and M. Cashel, 1988. Basal ppGpp level adjustment shown by new spoT mutants affect steady state growth rates and rrnA ribosomal promoter reulation in Escherichia coli. Molecular and General Genetics 213:214–222.
- Cooper, V. S., A. F. Bennett, and R. E. Lenski, 2001. Evolution of thermal dependence of growth rate of Escherichia coli populations during 20,000 generations in a constant environment. Evolution 55:889–896.
- Cooper, V. S., and R. E. Lenski, 2001. The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407:736–739.
- Hastings, P. J., A. Slack, J. F. Petrosino, and S. M. Rosenberg, 2004. Adaptive amplification and point mutation are independent mechanisms: Evidence for various stress-inducible mutation mechanisms. PLoS Biology 2:2220–2233.
- Cairns, J., and P. L. Foster, 1991. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128:695–701.
- Cairns, J., J. Overbaugh, and S. Miller, 1988. The origin of mutants. Nature 335:142–145.
- Foster, P. L., 1999. Mechanisms of stationary phase mutation: A decade of adaptive mutation. Annual Review of Genetics 33:57–88.
- Cairns and Foster, Ref. 20.
- Harris, R. S., K. J. Ross, and S. M. Rosenberg, 1996. Opposing roles of the Holliday junction processing systems of Escherichia coli in recombination-dependent adaptive mutation. Genetics 142:681–691.
- McKenzie, G. J., R. S. Harris, P. L. Lee, and S. M. Rosenberg, 2000. The SOS response regulates adaptive mutation. Proceedings of the National Academy of Sciences, USA 97:6646–6651.
- Stumpf, J. D., A. R. Poteete, and P. L. Foster, 2007. Amplification of lac cannot account for adaptive mutation to Lac+ in Escherichia coli. Journal of Bacteriology 189:2291–2299.
- Kugelberg, E., E. Kofoid, A. B. Reams, D. I. Andersson, and J. R. Roth, 2006. Multiple pathways of selected gene amplication during adaptive mutation. Proceedings of the National Academy of Science, USA 103:17319–17324.
- Roth, J. E., E. Kugelberg, A. B. Reams, E. Kofoid, and D. I. Andersson, 2006. Origin of mutations under selection: The adaptive mutation controversy. Annual Reviews in Microbiology 60:477–501.
- Pettersson, M. E., D. I. Andersson, J. R. Roth, and O. G. Berg, 2005. The amplification model for adaptive mutation: Simulations and analysis. Genetics 169:1105–1115.
- Hall, B. G., 1990. Spontaneous point mutations that occur more often when advantageous than when neutral. Genetics 126:5–16.
- Torkelson, J., R. S. Harris, M.-J. Lombardo, J. Nagendran, C. Thulin, and S. M. Rosenberg, 1997. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO Journal 16: 3303–3311.
- Roth, J. R., E. Kofoid, F. P. Roth, O. G. Berg, J. Seger, and D. I. Andersson, 2003. Regulating general mutation rates: examination of the hypermutable state model for Cairnsian adaptive mutation. Genetics 163:1483–1496.
- Rosche, W. A., and P. L. Foster, 1999. The role of transient hypermutators in adaptive mutation in Escherichia coli. Proceedings of the National Academy of Science, USA 96:6862–6867.
- Helling, R. B., C. N. Vargas, and J. Adams, 1987. Evolution of Escherichia coli during growth in a constant environment. Genetics 116:349–358.
- Adams, J., 2004. Microbial evolution in laboratory environments. Research in Microbiology 155:311–318.
- Treves, D. S., S. Manning, and J. Adams, 1998. Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Molecular Biology and Evolution 15:789–797.
- Saumaa, S., A. Tover, L. Kasak, and M. Kivisaar, 2002. Different spectra of stationary-phase mutations in early-arising versus late-arising mutants of Pseudomonas putida: Involvement of the DNA repair enzyme MutY and the stationary-phase sigma factor RpoS. Journal of Bacteriology 184:6957–6965.
- Shu, J., H. E. Schellhorn, and T. M. Murphy, 2006. Stationary phase-induction of G→T mutations in Escherichia coli. Mutation Research. Fundamental and Molecular Mechanisms of Mutagenesis 596:106–112.
- Saumaa et al., Ref. 38.
- Koorits, L., R. Tegova, M. Turk, K. Tarassova, A. Tover, and M. Kivisaar, 2007. Study of the involvement of ImuB and DnaE2 in stationary-phase mutatgenesis in Pseudomonas putida. DNA Repair 6:863–868.
- Adams, Ref. 36.
- Bull, H. J., M.-J. Lombardo, and S. M. Rosenberg, 2001. Stationary-phase mutation in the bacterial chromosome: recombination protein and DNA polymermase IV dependence. Proceedings of the National Academy of Science, USA 98:8334–8341.
- Harris, R. S., G. Fend, K. J. Ross, R. Sidhu, C. Thulin, S. Longerich, S. K. Szigety, M. E., Winkler, and S. M. Rosenberg, 1997. Mismatch repair protein MutL becomes limiting during stationary-phase mutations. Genes and Development 11: 2426–2437.
- Pedraza-Reyes, M., and R. E. Yasbin, 2004. Contribution of the mismatch DNA repair system to the generation of stationaryphase- induce mutants of Bacillus subtilis. Journal of Bacteriology 186(19):6485–6491.
- Bjedov, I., O. Tenaillon, B. Gérard, V. Souza, E. Denamur, M. Radman, F. Taddel, and I. Matic, 2003. Stress-induced mutagenesis in bacteria. Science 300:1404–1409.
- Zambrano, M. M., D. Siegele, M. Almirón, A. Tormo, and R. Kolter, 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259:1757–1760.
- Farrell, M. J., and S. E. Finkel, 2003. The growth advantage in stationary-phase phenotype conferred by rpoS mutations is dependent on the pH and the nutrient environment. Journal of Bacteriology 185:7044–7052.
- Zinser, E., and R. Kolter, 2000. Prolonged stationary-phase incubation selects for lrp mutations in Escherichia coli K-12. Journal of Bacteriology 182:4361–4365.
- Calvo, J. M., and R. G. Matthews, 1994. The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiology Research 58:466–490.
- Zinser and Kolter, Ref. 53.
- Zinser, E., and R. Kolter, 2004. Escherichia coli evolution during stationary phase. Research in Microbiology 155:328–336.
- Somerville, G. A., M. S. Chaussee, C. I. Morgan, J. R. Fitzgarald, D. W. Dorwad, L. J. Reitzer, and J. M. Musser, 2002. Staphylococcus aureus aconitase inactivation unexpectedly inhibits post-exponential-phase growth and enhances stationaryphase survival. Infection and Immunity 70:6373–6382.
- Zinser, E., and Kolter, R. (1999) Mutations enhancing amino acid catabolism confer a growth advantage in stationary phase. Journal of Bacteriology 181:5800–5807.
- Zinser, E., D. Schneider, M. Blot, and R. Kolter, 2003. Bacterial evolution through the selective loss of beneficial genes: Tradeoffs in expression involving two loci. Genetics 164:1271–1277.
- Schultz, J. E., and A. Matin, 1991. Molecular and functional characteristics of a carbon starvation gene of Escherichia coli. Journal of Molecular Biology 218:129–140.
- Zinser et al., Ref. 59.
- Zinser and Kolter, Ref. 56.
- Riehle, M. M., A. F. Bennett, and A. D. Long, 2001. Genetic architecture of thermal adaptation in Escherichia coli. Proceedings of the National Academy of Sciences, USA 98:525–530.
- Riehle, Bennett, and Long, Ref. 66.
- Kugelberg, et al., Ref. 28.
- Roth, J. R., N. Benson, T. Galitski, K. Haack, J. G. Lawrence, and L. Miesel, 1996. In Niedhardt, F. C. (ed)., Escherichia coli and Salmonella cellular and molecular biology, Vol. 2. Washington, DC: ASM Press, pp. 2256–2276.
- Riehle, M. M., A. F. Bennett, R. E. Lenski, and A. D. Long, 2003. Evolutionary changes in heat-inducible gene expression in lines of Escherichia coli adapted to high temperature. Physiological Genomics 14:47–58.
- Riehle, M. M., A. F. Bennett, and A. D. Long, 2005. Changes in gene expression following high-temperature adaptation in experimentally evolved populations of E. coli. Physiological and Biochemical Zoology 78:299–315.
- Anderson, K. L. (2005). Is bacterial resistance to antibiotics an appropriate example of evolutionary change? Creation Research Society Quarterly 41:318–326.
- Anderson, Ref. 74.
- Miller, C., L. E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer, and S. N. Cohen, 2004. SOS response induction by ß-lactams and bacterial defense against antibiotic lethality. Science 305:1629–1631.
- Paul, R., S. Postius, K. Melchers, and K. P. Schäfer, 2001. Mutations of the Helicobacter pylori genes rdxA and pbp1 cause resistance against metronidazole and amoxicillin. Antimicrobial Agents and Chemotherapy 45:962–965.
- Kashiwagi, K., M. H. Tsuhako, K. Sakata, T. Saisho, A. Igarashi, S. O. P. DaCosta, and K. Igarashi, 1998. Relationship between spontaneous aminoglycoside resistance in Escherichia coli and a decrease in oligopeptide binding protein. Journal of Bacteriology 180:5484–5488.
- Thwaites, W. M., 1985. New proteins without God’s help. Creation/Evolution 16:1–3.
- Yasuhira, K., Y. Tanaka, H. Shibata, Y. Kawashima, A. Ohara, D. Kato, M. Takeo, and S. Negoro, 2007. 6-Aminohexanoate oligomer hydrolases from the alkalophilic bacteria Agromyces sp. Strain KY5R and Kocuria sp. Strain KY2. Applied and Environmental Microbiology 73:7099–7102.
- Negoro, S., 2000. Biodegradation of nylon oligomers. Applied Microbiology and Biotechnology 54:461–466.
- Yasuhira, et al., Ref. 80.
- Yasuhira, K., Y. Uedo, N. Shibata, S. Negoro, M. Takeo, and Y. Higuchi, 2006. Crystallization of X-ray diffraction analysis of 6-aminohexanoate-cyclic-dimer hydrolase from Arthrobacter sp. K172. Acta Crystallographic F62:1209–1211.
- Negoro, S., T. Ohki, N. Shibata, N. Mizuno, Y. Wakitani, J. Tsurukame, K. Matsumoto, I. Kawamoto, M. Takeo, and Y. Higuchi, 2005. X-ray cystallographic analysis of 6-aminohexanoate-dimer hydrolase. Journal of Biological Chemistry 280: 39644–39652.
- Ohki, T., Y. Wakitani, M. Takeo, K. Yasuhira, N. Shibatat, Y. Higuchi, and S. Negoro, 2006. Mutational analysis of 6- aminohexanoate-dimer hydrolase: Relationship between nylon oligomer hydrolytic and esterolytic activities. FEBS Letters 580:5054–5058.
- Negoro, S., T. Ohki, N. Shibata, K. Sasa, H. Hayashi, H. Nakano, K. Yasuira, D. Kato, M. Takeo, and Y. Higuchi, 2007. Nylon-oligomer degrading enzyme/substrate complex: Catalytic mechanism of 6-aminohexanoate-dimer hydrolase. Journal of Molecular Biology 370:142–156.
- Fersht, A. R., 1985. Enzyme structure and mechanism. San Francisco, California: Freeman Press.
- Bone, R., J. L. Silen, and D. A. Agard, 1989. Structural plasticity broadens the specificity of an engineered protease. Nature 339:191–195.
- Negoro, Ref. 81.
- Gautam, R., A. S. Bassi, and E. K. Yanful, 2007. A review of biodegradation of synthetic plastic and foams. Applied Biochemistry and Biotechnology 141:85–108.
- Anderson, K. L., and A. A. Salyers, 1989. Genetic evidence that outer membrane binding is required for starch utilization by Bacteroides thetaiotaomicron. Journal of Bacteriology 171:3199–3204.
- Bushman, F., 2002. Laterial DNA transfer. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press.
- Bushman, Ref. 95.
- Snyder and Champness, Ref. 1.
- Bennett, A. F., and R. E. Lenski, 2007. An experimental test of evolutionary trade-offs during temperature adaptation. Proceedings of the National Academy of Sciences, USA 104:8649–8654.
- Cullum, A. J., A. F. Bennett, and R. E. Lenski, 2001.Evolutionary adaptation to temperature. IX. Preadaption to novel stressful environments of Escherichia coli adapted to high temperature. Evolution 55:2194–2202.
- Frair, W., 2000. Baraminology—classification of created organisms. Creation Research Society Quarterly 37:82–91.
- Friedmann, H. C., 2004. From “Butyribacterium” to “E. Coli.” Perspectives in Biology and Medicine 47:47–66.
- Purdom, G., and K. Anderson, 2008. Analysis of Barry Hall’s research of the E. coli ebg Operon: Understanding the Implications for bacterial adaptation to adverse environments. In Snelling, A. A. (ed.), Proceedings of the sixth international conference on creationism. Pittsburgh, Pennsylvania: Creation Science Fellowship and Dallas, Texas: Institute for Creation Research, pp. 149–163.
- Pál, C., B. Papp, M. J. Lercher, P. Csermely, S. G. Oliver, and L. D. Hurst, 2006. Chance and necessity in the evolution of minimal metabolic networks. Nature 440:667–670.
- Anderson, K. L., 2000. Degradation of cellulose and starch by anaerobic bacteria. In Doyle, R. J. (ed.), Glycomicrobiology. New York, New York: Kluwer Academic/Plenum Publishers, pp. 359–386.
- Sanford, J., 2005. Genetic entropy & the mystery of the genome. Lima, New York, New York: Ivan Press.
- Sanford, Ref. 106.
- ENCODE Project Consortium. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816.
- Rigoutsos, I., T. Huynh, K. Miranda, A. Tsirigos, A. McHardy, and D. Platt, 2006. Short blocks from the noncoding parts of the human genome have instances within nearly all known genes and relate to biological processes. Proceedings of the National Academy of Sciences 103:6605–6610.
- Bertone, P., V. Stolc, T. E. Royce, J. S. Rozowsky, A. E. Urban, X. Zhu, J. L. Rinn, W. Tongprasit, M. Samanta, S. Weissman, M. Gerstein, and M. Snyder, 2004. Global identification of human transcribed sequences with genome tiling arrays. Science 306:2242–2246.
- ReMine, W., 2006. More precise calculations of the cost of substitution. Creation Research Society Quarterly 43: 111–120.
- Loeb, L. A., 1991. Mutator phenotype may be required for multistage carcinogenesis. Cancer Research 51:3075–3079.
- Hall, B. G., 1995. Adaptive mutations in Escherichia coli as a mode for the multiple mutational origins of tumors. Proceedings of the National Academy of Science, USA 92:5669–5673.
- Loeb, Ref. 115.
- Richards, B., H. Zhang, G. Phear, and M. Meuth, 1997. Conditional mutator phenotypes in hMSH2-deficient tumor cell lines. Science 277:1523–1526.
- Hall, Ref. 116.
Support the creation/gospel message by donating or getting involved!

Answers in Genesis is an apologetics ministry, dedicated to helping Christians defend their faith and proclaim the good news of Jesus Christ.
- Customer Service 800.778.3390
- Available Monday–Friday | 9 AM–5 PM ET
- © 2026 Answers in Genesis