
A Brief History of Intolerance in Modern Cosmology
pp. 1–9 • Dr. Jerry Bergman
A review of some recent well-documented cases of intolerance in the cosmology field illustrates a common problem in science. Many relate to the Big Bang theory.
MLACopy
APACopy
Chicago Copy
${currentCitationStyle} Citation Copied to Clipboard
pp. 1–9 • Dr. Jerry Bergman
A review of some recent well-documented cases of intolerance in the cosmology field illustrates a common problem in science. Many relate to the Big Bang theory.
pp. 11–20 • Todd Charles Wood
Some contend that Charles Darwin plagiarized his theory of evolution. Whether you agree with the man or not, however, the evidence suggests the idea is his.

pp. 21–28 • Tom Hennigan
As we seek to discover the intent of the Designer, we can enhance our stewardship of the land by using this symbiosis to re-establish polluted and disturbed landscapes and grow sustainable crops.

pp. 29–51 • John Matthews
Certain features of the “Upper Cretaceous” period correspond closely with the biblical account of the Noachian Flood around day 150, and uniformitarian explanations for “chalk” are inadequate.

pp. 53–78 • Dr. Andrew A. Snelling , et. al.
Polonium radiohalos found in biotite flakes of granites in Yosemite National Park place severe time constraints on the formation and cooling of the granite plutons.

pp. 79–84 • Dr. Jean Lightner
Recognizing the population bottleneck which occurred in land animals at the time of the Flood, it is clear that genetic variation was once more limited than it is today.
pp. 85–95 • Dr. Georgia Purdom , et. al.
Microbes form a life-sustaining organosubstrate on earth and contribute to our understanding of geology, ecology, and biology.

pp. 97–106 • Yingguang Liu , et. al.
Evolutionists assume that all endogenous retroviruses are remnants of germ line infection by exogenous retroviruses.
pp. 107–115 • Daniel Criswell
It is apparent from the knowledge gained about mitochondria ribosome structure and function since the proposal of the Serial Endosymbiosis Theory that prokaryotes are not the ancestors of eukaryotes.
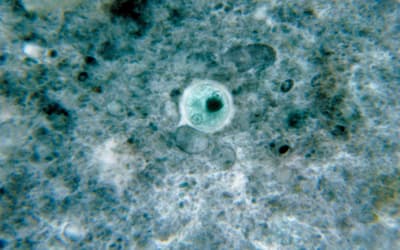
pp. 117–121 • Frank Sherwin
There is a need for parasites such as Entamoeba histolytica to be addressed from a biblical perspective that may include their original symbiotic or mutualistic association in man.

pp. 123–131 • Ira Loucks
Fungi are intriguing organisms with a wealth of diversity in their morphology and ecology. Determining the fundamentals of their biology from a biblical perspective is a daunting but achievable task.
pp. 133–150 • Dr. Georgia Purdom
Bacteria are mostly beneficial, even though a minority are known as pathogens. They are necessary for natural processes such as human digestion and biogeochemical cycling.
pp. 151–167 • Dr. Terry Mortenson
The problem of evil is always a challenge for the Christian witness. However, William Dembski’s “solution” proves logically and biblically unsound.
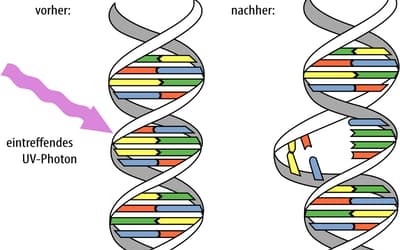
pp. 169–174 • Jonathan Bartlett
Mutations are normally classified according to their proximal effect on an organism’s fitness, whether beneficial, deleterious, or neutral.
pp. 175–200 • Dr. Terry Mortenson
In the past few decades there has been a growing controversy in society and in the Church over evolution and the age of the earth.
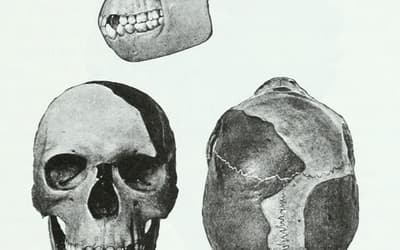
pp. 201–210 • Dr. Jerry Bergman
A review of the history of paleoanthropology leads to the conclusion that the discipline is far less objective than that for physics, chemistry, or even biology.